
Folate Transport into the Nervous System
It is remarkable and unusual that cerebrospinal fluid (CSF) folate levels in man are approximately three times higher than in serum. In epileptic, neurological and psychiatric patients we confirmed a close correlation between serum and CSF folate levels, suggestive of an active transport mechanism for the vitamin into the nervous system. With I. Chanarin we demonstrated that methyl folate is the form of the vitamin exclusively transported across the Blood Brain Barrier. Furthermore during treatment with folic acid CSF folate levels rose only very slowly (in contrast to serum) due to the active barrier mechanism limiting the rate of entry of such an excitatory compound (see Section on Epilepsy).
Key references:
Reynolds E.H., Gallagher B.B., Mattson R.H., Bowers M., Johnson A.L. Relationship between serum and cerebrospinal fluid folate. Nature 1972; 240: 155-157
Chanarin I., Perry J., Reynolds E.H. Transport of 5-methyltetrahydrofolic acid into the cerebrospinal fluid in man. Clin Sci Mol Med 1974; 46: 369-373
Folate and Neuropsychiatry Conference

By the late 1970s there was growing interest and evidence of a neuropsychiatry of folate deficiency, reinforced by recent studies by Botez and colleagues in Montreal. M. Botez and I therefore convened an International Symposium in Montreal in 1978 at which the concept of a neuropsychiatry of folate deficiency was now firmly established.
Key reference:
Botez M.I., Reynolds E.H., Editors. Folic Acid in Neurology, Psychiatry and Internal Medicine. Raven Press, New York, 1979
Folate, S-Adenosylmethionine (SAM), Methylation and Mood
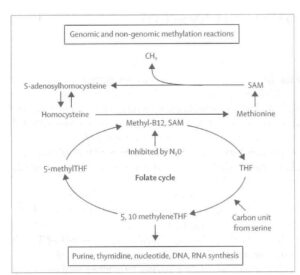
At this time I learnt of Italian studies suggesting that SAM had antidepressant properties in patients with endogenous depression. Knowing the links between the folate cycle (the source of methyl groups) and the methylation cycle (figure) in which SAM is the methyl donor in innumerable methylation reactions involving eg nucleoproteins, proteins, phospholipids, neurotransmitters and monoamlines, it seemed too much of a coincidence that folate deficiency could lead to depression and SAM had antidepressant properties. This suggested to me that methylation processes in the brain were implicated in disorders of mood and perhaps cognitive decline.
In clinical trials of intravenous and oral SAM in collaboration with M. Carney at Northwick Park we confirmed the antidepressant potential of SAM, especially in bipolar depression, in which 9 of 11 patients switched into euphoria or hypomania, and to a lesser extent in endogenous depression. We also showed that CSF SAM levels rose with SAM treatment, and this was associated with an increase in serotonergic and dopaminergic activity, the latter having also been reported by Botez and colleagues in association with folate therapy for depression.
We also reported that CSF SAM levels were significantly lower in depression, but they were even lower in patients with dementia, including Alzheimer’s Disease.
In the 1990s several large epidemiological studies were now suggesting that high homocysteine levels (see figure), low folate levels and sometimes low serum vitamin B12, were independent risk factors for dementia. My colleagues and I also found that homocysteine levels were raised in approximately half of patients with severe depression. We now identified a subgroup of patients with severe depression associated with raised plasma homocysteine; low serum, red cell and CSF folate; and low CSF SAM and monoamine metabolites.
Key references:
Reynolds E.H., Carney M.W.P., Toone B.K. Methylation and mood. Lancet 1984; 2: 196-198
Bottiglieri T., Laundy M., Crellin R., Toone B.K., Carney M.W,P., Reynolds E.H. Homocysteine, folate, methylation and monoamine metabolism in depression. J.Neurol Neurosurg Psychiat 2000; 69: 228-232
Reynolds E.H. Folic acid, ageing, depression and dementia. BMJ 2002; 324: 1512-1515
Treatment
Patients presenting with the neuropsychiatric associations of megaloblastic anaemia, whether due to folate or vitamin B12 deficiency, usually respond well to the appropriate vitamin therapy unless the disease has been prolonged and severe, when the response is often partial.
Less certain is the significance of folate deficiency in neuropsychiatric disorders in the absence of anaemia. In a placebo-controlled trial my colleague A. Coppen reported that a small dose of folic acid for one year enhanced recovery from bipolar depression in patients on lithium. In a placebo-controlled trial of methyl folate in addition to standard psychotropic medication in psychiatric admissions with folate deficiency (red cell folate less than 200 ng/ml) my colleagues and I reported significant improvement in clinical and social recovery in both major depression and schizophrenia. The clinical and biological evidence suggest that the response to the vitamin therapy is usually slow, requiring up to 6 months for optimum benefit, bearing in mind the Blood Brain Barrier mechanisms discussed above.
More recently my colleagues and I reported a pilot trial of methyl folate against amitriptyline as monotherapy for mild to moderate depression in outpatients for 6 weeks. The most striking finding was that responders to the vitamin exhibited a much greater rise in red cell folate than non-responders. The numbers were small and the differences between the vitamin and a standard antidepressant were not significant, but a large scale trial would be justified.
Key references:
Godfrey P.S.A., Toone B.K., Carney M.W.P., Flynn T.G, Bottiglieri T., Laundy M., Chanarin I., Reynolds E.H. Enhancement of recovery from psychiatric illness by methylfolate. Lancet 1990; 2: 392-395
Reynolds E.H. Neurometabolic approach to treatment-resistant depression. Brit J Psychiat 2019; 215: 568-569
Neural Tube Defects, Folate Metabolism and Food Fortification
In the late 1980s and early 1990s evidence emerged that folate deficiency could increase the risks of neural tube defects (NTDs) and that small doses of the vitamin prior to and in the early stages of pregnancy could reduce the risk of NTDs by approximately 50%. This confirmed the importance of folate metabolism in foetal and child development. In the early 2000s many governments decided to fortify bread or cereals with folic acid as a public health policy to reduce the risks.
I have spent much time in the last 20 years reminding others of the risks of excess folate to the nervous system, which was well documented in the past, but which advocates of fortification have been inclined to doubt, dismiss or ignore. I was a member of the UK Committee on Medical Aspects of Food and Nutrition Policy (COMA) in 2000 and was a lone voice in opposing fortification because of the potential risks to nervous system function, especially cognition, in a much larger elderly population who are additionally vulnerable because of increasing incipient vitamin B12 deficiency. Although most European countries, including the UK, have held back, about 80 countries worldwide have implemented fortification policies. Although the modest benefits of fortification are undoubted, as I and others have pointed out there is already epidemiological evidence of harm to cognition in the post-fortification era, reinforced by metabolic studies of the complex interaction of folate and vitamin B12 metabolism. There is now experimental evidence of a biphasic response to the vitamin in nerve regeneration or cortical development in which an optimum dose response is followed by reversal with increasing doses. This remains a contentious issue.
Key references:
Reynolds E.H. Benefits and risks of folic acid to the nervous system. J Neurol Neurosurg Psychiat 2002; 72: 567-571
Reynolds E.H. What is the safe upper intake level of folic acid for the nervous system? Implications for folic acid fortification policies. Europ J Clin Nutr 2016; 70: 537-540
Reynolds E.H. The risks of folic acid to the nervous system in vitamin B12 deficiency: rediscovered in the era of folic acid fortification policies. J Neurol Neurosurg Psychiat 2017; 88: 1097-1098
Subacute Combined Degeneration with high VitaminB12: a new Syndrome
SCD occurs more commonly with vitamin B12 deficiency than folate deficiency. In the former deficiency SCD is invariably associated with some of the lowest serum vitamin B12 levels and, interestingly, some of the highest serum folate levels.
My colleagues and I have described a new syndrome or ‘ a new cause of an old syndrome’ in which SCD was associated with a very high serum vitamin B12, but tissue vitamin B12 deficiency due to an abnormal plasma vitamin B12 binding protein. The SCD may have been aggravated by inappropriate treatment with folic acid due to misdiagnosis because of the high serum vitamin B12, but the patient recovered fully with vitamin B12 treatment. Serum vitamin B12 levels are not always a reliable guide to vitamin B12 status.
Key references:
Reynolds E.H., Bottiglieri T., Laundy M., Stern J., Payan J., Linnell J., Faludy J. Sub-acute combined degeneration with high serum vitamin B12 level and abnormal vitamin B12 binding protein. New cause of an old syndrome. Arch Neurol 1993;50: 739-742.
Multiple Sclerosis (MS) and Vitamin B12 Deficiency
MS is usually clinically and pathologically clearly distinguishable from the neurology of vitamin B12 deficiency, although demyelination is a prominent feature of both conditions. Furthermore vitamin B12 deficiency is a disorder of middle and later life and is relatively uncommon in the younger age range of presentation of MS. Nevertheless I and my colleagues reported 10 cases of this association in 1991. The cases were unusual in that the cause of the vitamin B12 deficiency was unclear in most, only 2 having pernicious anaemia. A disorder of vitamin B12 binding or transport was suspected. The clinical features were typical of MS and, interestingly, although severe vitamin B12 deficiency was present none of the patients had peripheral neuropathy, which is a common feature of the neurology of vitamin B12 deficiency, but not MS.
We therefore undertook further studies of vitamin B12 metabolism in a series of unselected patients with MS and reported a significant increase of mean cell volume (MCV), associated with a decline in serum vitamin B12 and elevation of plasma homocysteine levels compared with matched controls. Vitamin B12 deficiency is more common in MS than previously suspected but only a minority of patients exhibit this association.
The significance of the association of MS and vitamin B12 deficiency remains uncertain. Could it represent an overlap between two autoimmune diseases that have some similarities in geographical and sex ratio distribution? Do the immune reactions and repair processes in MS increase the demand for vitamin B12? Could the vitamin deficiency play a causal or aggravating role in some patients with MS? There are arguments against all these interpretations, not least because the cause of vitamin B12 deficiency is unclear in most of these cases. Many of this subgroup of MS patients and some others without vitamin B12 deficiency claim that vitamin B12 therapy improves their symptoms and wellbeing, but further studies are required to clarify this unusual association.
Key references:
Reynolds E.H., Linnell J.C., Faludy J.E. Multiple sclerosis associated with vitamin B12 deficiency. Arch Neurol 1991; 48: 808-811.
Reynolds E.H. Multiple sclerosis and vitamin B12 metabolism. J Neurol Neurosurg Psychiat 1992; 50: 339-340
